A propos de deux cas de syndrome auto-immun multiple. Une entité à connaitre
Z. Chikh Salah, F. Otmani*, N. Oumnia
Service de médecine interne, EHS Salim Zemirli,
*Centre hospitalo-universitaire Mustapha Bacha, Alger, Algérie.
RÉSUMÉ
Le syndrome auto-immun multiple (SAIM) est défini par l’apparition, la survenue simultanée ou successive, chez un même patient d’au moins trois maladies auto- immunes, spécifiques et non spécifiques d’organe, ce qui fait la particularité de cette entité rare. Il existe trois types de SAIM en fonction des maladies associées: Le SAIM de type 1 regroupe la myasthénie auto-immune, les thymomes, la polymyosite et la myocardite auto-immune. Le SAIM de type 2 comprend le syndrome de Gougerot-Sjögren, la polyarthrite rhumatoïde, la cirrhose biliaire primitive, la sclérodermie et les thyroïdites auto-immunes. Le SAIM de type 3 contient les thyroïdites auto-immunes, la myasthénie, les thymomes, le syndrome de Gougerot Sjögren, la maladie de Biermer, le purpura thrombopénique idiopathique, la maladie d’Addison, le diabète de type 1, le vitiligo, l’anémie hémolytique auto-immune, le lupus et la dermatite herpétiforme. Nous rapportons le premier cas de madame F. D. âgée de 68 ans, aux antécédents de thyroïdite auto immune évoluant depuis 03 ans, d’un phénomène de Raynaud et d’un syndrome sec évoluant depuis 02 ans où le diagnostic d’ une sclérodermie dans sa forme cutanée systémique limité est évoqué, associé à une cirrhose biliaire primitive découverte suite à une exploration d’ un syndrome de cholestase anictérique. Il s’agit d’un SAIM de type 2. Le deuxième cas, c’est madame A. F. âgée de 43 ans, au antécédent d’un diabète de type I depuis l’enfance, hospitalisée pour exploration d’une anémie sévère où le diagnostic d’une anémie de Biermer est retenu associé à une thyroïdite auto immune d’Hashimoto et un syndrome de gougerot Sjögren puis, réhospitalisée pour exploration d’ une cholestase anéctérique révélant une hépatite auto immune type 1. Il s’agit d’un SAIM de type 3.
Mots clés : Syndrome auto-immun multiple, désordres auto-immuns, rare.
SUMMARY :
Multiple autoimmune syndrome (MIAS) is defined by the appearance, the simultaneous or successive occurrence, in the same patient, of at least three autoimmune diseases, specific and non-specific for the organ, which makes the peculiarity of this rare entity. There are three types of SAIM depending on the associated diseases: SAIM type 1 includes autoimmune myasthenia, thymomas, polymyositis and autoimmune myocarditis. SAIM type 2 includes Gougerot Sjögren syndrome, rheumatoid arthritis, primary biliary cirrhosis, scleroderma and autoimmune thyroiditis. SAIM type 3 contains autoimmune thyroiditis, myasthenia gravis, thymomas, Gougerot Sjögren’s syndrome, Biermer’s disease, idiopathic thrombocytopenic purpura, Addison’s disease, type 1 diabetes, vitiligo, l autoimmune hemolytic anemia, lupus and dermatitis herpetiformis. We report the first case of Mrs. F. D. aged 68, with a history of autoimmune thyroiditis evolving for 03 years, Raynaud’s phenomenon and dry syndrome evolving for 02 years where the diagnosis of scleroderma in its form Limited systemic skin is evoked, associated with primary biliary cirrhosis discovered following an exploration of a syndrome of anicteric cholestasis. This is a SAIM type 2. The second case is Mrs. A. F. aged 43, with a history of type I diabetes since childhood, hospitalized for exploration of severe anemia where the Diagnosis of Biermer’s anemia is retained associated with Hashimoto’s autoimmune thyroiditis and Sjögren’s gougerot syndrome then, rehospitalized for exploration of anecteric cholestasis revealing autoimmune hepatitis type 1. It is a SAIM type 3.
Key Word : Multiple autoimmune syndrome, auto-immune desorder, rare
INTRODUCTION
Les syndromes auto-immuns multiples, groupe hétérogène de maladies impliquant des désordres auto-immuns, réalisent une condition pathologique rare, décrite en 1988 par Humbert et Dupond (1, 2). Il en existe quatre groupes (3). Le type 3, le plus fréquent, est lui-même divisé en quatre sous-groupes en fonction du type d’organe atteint : l’appareil gastro-intestinal dans le type 3B, la peau, le système hématopoïétique ou nerveux central dans le type 3C. Une maladie thyroïdienne auto-immune est toujours associée (4, 5). La survenue simultanée ou successive, chez un même individu, de trois maladies auto-immunes ou plus, est reconnue sous le terme de Syndrome Auto-immun Multiple (SAIM). Nous rapportons deux observations particulières par la succession de plusieurs maladies sur une période de 10 ans (3, 6).
OBSERVATION 1
La première observation rapporte le cas d’un syndrome auto-immun multiple de type 2, comprenant une cirrhose biliaire primitive, une sclérodermie systémique cutanée limitée et une thyroïdite auto immune. La patiente F. D. âgée de 68 ans, aux antécédents d’hypothyroïdie sous Lévothyrox (75mg/j) depuis 03 ans, en euthyroïdie et de diabète type 1 chez sa fille. Admise pour prise en charge d’une dyspnée d’effort stade 2 évoluant depuis 2 ans et d’un syndrome de choléstase anictérique. Cette dyspnée rentre dans le cadre d’une sclérodermie systémique cutanée limitée, sur les éléments suivant:
Clinique :
Dyspnée d’effort stade 2.
Une toux sèche.
Des râles crépitant aux deux bases pulmonaires.
Un phénomène de Raynaud aux extrémités.
Une Sclérodactylie.
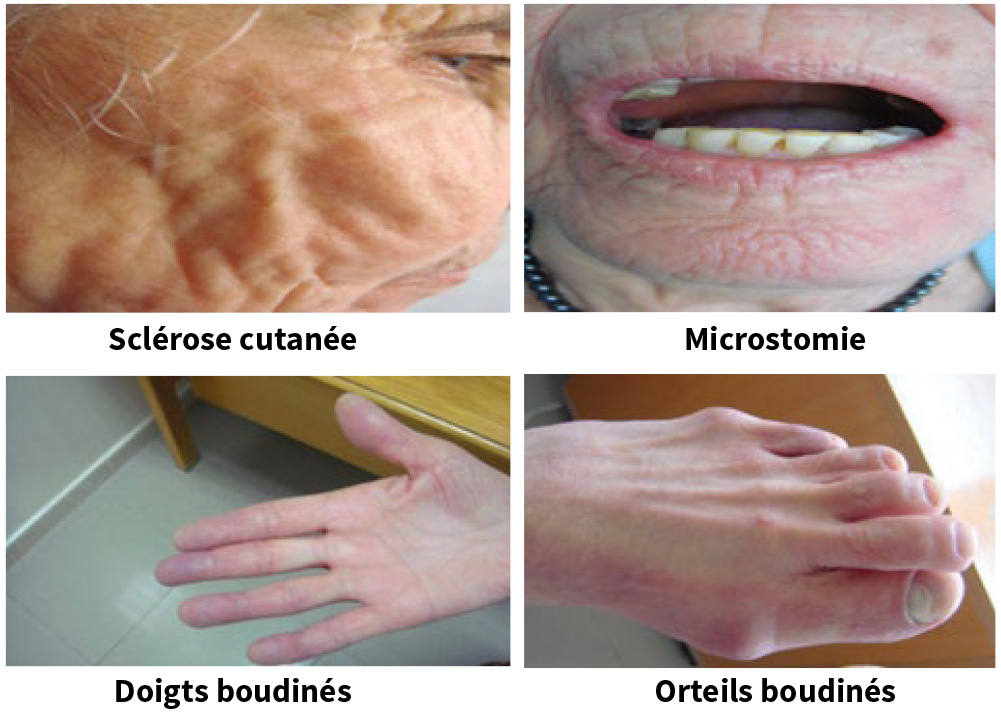
Sclérose cutanée distale du visage, des pieds, des mains avec doigts boudinés
et un Rodnan à 07.
Une peau fine cartonnée avec accentuation des rides.
Une microstomie (limitation de l’ouverture de la bouche à 3 cm)
Un syndrome sec : buccal, oculaire et génital.
Examens biologiques :
Syndrome inflammatoire :
Hyper gammaglobulinémie poly clonale à 25 g/l et une VS accélérée à 78/90
mm.
Bilans immunologique: Les AC anti Scl 70, les AC anti-Jo1, les AC anti-nucléaire,
les AC anti-SSA et les AC anti-SSB sont négatifs. Les AC anti-DNA sont positifs
à 79 UI/l.
Examens morphologiques:
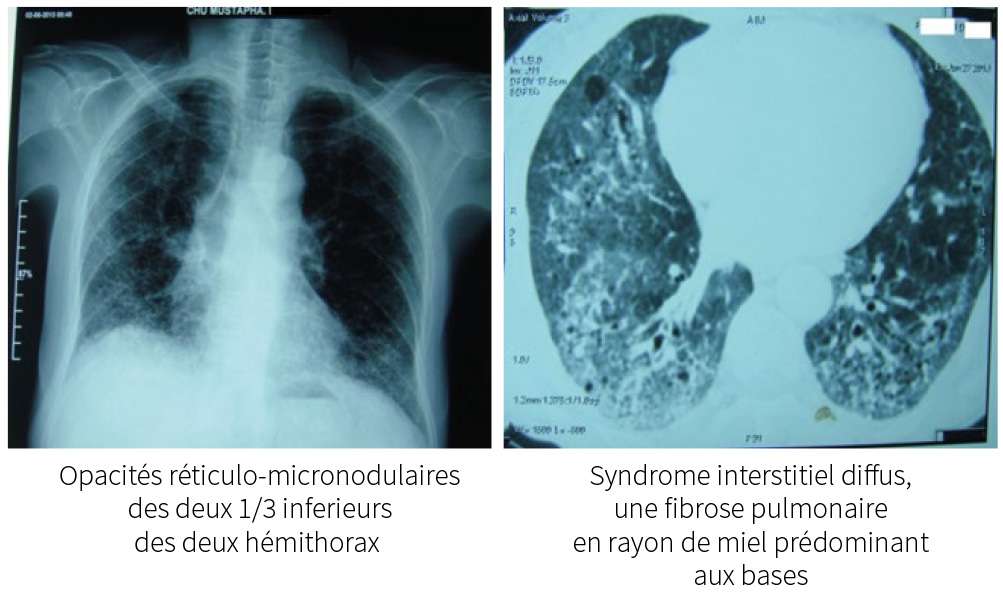
Radiologie pulmonaire : opacités réticulo-micronodulaires des deux 1/3 inferieurs
des deux hémi thorax.
TDM thoracique : syndrome interstitiel diffus, une fibrose pulmonaire en rayon
de miel prédominant aux bases.
L’épreuve fonctionnelle respiratoire : trouble ventilatoire mixte à prédominance
restrictive, CPT 28%, CVF 57%, VEMS 61%.
La bronchoscopie : muqueuse bronchique inflammatoire et élargissement des
éperons inter-lobaire.
L’étude du LBA : est sans particularité.
La biopsie des éperons bronchiques : le chorion bronchique retrouve des
glandes régulières et un discret infiltrat inflammatoire non spécifique.
La capillaroscopie : aspect de micro angiopathie avec présence de méga capillaires.
Diagnostic de la sclérodermie systémique cutanée limitée est évoqué selon les critères de classification de Le Roy et Medsger:
Clinique :
Un syndrome de Raynaud Une sclérose cutanée avec doigts boudinés et un score de Radnon à 07 Une microstomie
Examens morphologiques :
Présence des mégas capillaires à la capillaroscopie
Fibrose pulmonaire diffuse
Avec un syndrome restrictif à l’EFR sans retentissement cardiaque.
Examens biologiques :
Les AC anti Scl70 sont négatifs
Le syndrome de Gougerot Sjögren est suspecté cliniquement et écarté histologiquement:
Cliniquement
Un syndrome sec : buccal, oculaire et génital.
Uvéite antérieure chronique avec cataracte sénile débutante
Biopsie des glandes salivaires accessoires
Glandes salivaires de morphologie conservés, avec discrets remaniements fibro-inflammatoires.
Le diagnostic de thyroïdite chronique auto-immune est retenu selon :
La clinique
Signes d’hypothyroïdie associant une constipation, cheveux fins, frilosité
La biologie
TSHus, FT4 : hypothyroïdie primaire. AC anti-TPO, anti-Tg sont négatifs.
L’échographie cervicale
Une glande thyroïdienne de taille réduite, au contour régulier, d’écho structure finement remaniée, hypoéchogène sans nodule décelable évoquant une thyroïdite auto immune chronique.
La cholestase hépatique anictérique, sans obstacle extra-hépatique, évoluant depuis 2 ans est rattaché à une cirrhose biliaire primitive, devant:
La clinique
Uvéite antérieure bilatérale chronique.
Les examens biologiques
Cholestase hépatique anictérique avec des ɣGT 441UI/l (7xN) et des PAL 553
UI/l (3xN).
Cytolyse hépatique modérée avec ASAT 110 UI/l (3xN), ALAT 85 UI/l (2xN).
AC anti-nucléaire négatif
AC anti-mitochondrie à 1/1280 UI (32xN) de type M2.
AC anti-muscle lisse à 1/80 (2xN).
Les examens morphologiques
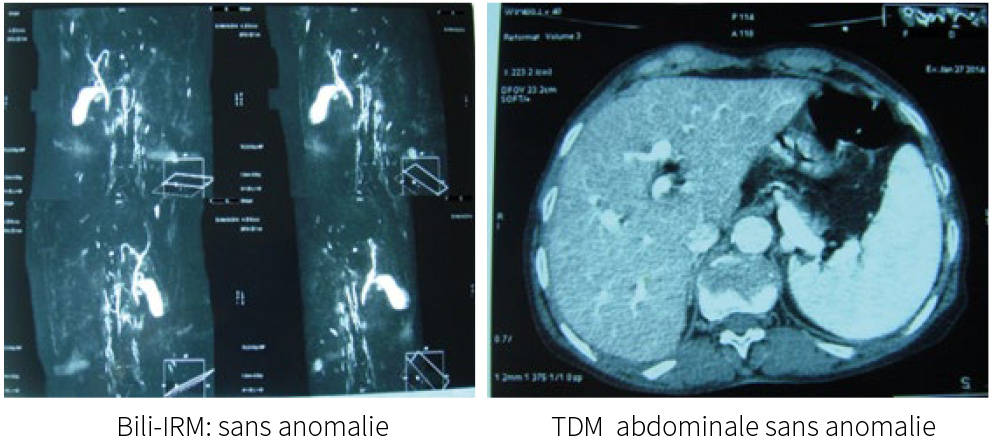
L’échographie, le scanner abdominal ainsi que la bili-IRM ne révèlent pas d’anomalie des voies biliaires intra et extra-hépatique ni du tissu hépatique.
L’examen histologique
Ponction biopsie hépatique: parenchyme hépatique d’architecture lobulée remaniée
par une fibrose portale, péri portale et septale avec mise en évidence
d’un infiltrat inflammatoire, évoquant une hépatopathie auto immune type
cirrhose biliaire primitive.
La ponction biopsie du foie a permis d’orienter le diagnostic vers une cirrhose
biliaire primitive et d’écarter l’association de celle-ci à une hépatite auto immune,
réalisant un overlap syndrome. L’association sclérodermie et Cirrhose
biliaire primitive rentre dans le cadre d’un syndrome de Reynolds. Un traitement
sous urso-désoxycholique est justifié.
OBSERVATION 2
La seconde observation rapporte le cas d’un syndrome auto-immun multiple de type 3c, associant diabète sucré de type 1, thyroïdite auto immune d’ Hashimoto, anémie de biermer, syndrome de Gougerot Sjögren et hépatite auto-immune de type 1. Mme A. F. âgée de 43 ans, aux antécédents d’un diabète sucré de type 1 et d’une hypothyroïdie sous traitement hormonale, hospitalisée pour la première fois pour une prise en charge d’un syndrome anémique sévère.
Le diagnostic d’une anémie de Biermer a été évoqué devant :
La clinique :
Anémie d’installation progressive avec pâleur, asthénie, dyspnée d’effort
d’aggravation progressive depuis six mois, et souffle systolique 2/6 méso cardiaque.
Troubles des phanères.
Un subictère, hépato-splénomégalie sans signe d’hypertension portale ni
signes d’appels digestifs.
Une langue lisse dépapillée (glossite).
Un syndrome pyramidal bilatéral (signe de babinski bilatéral)
Un syndrome cordonal postérieur (ataxie, paresthésies, aréflexie tendineuse,
troubles de la sensibilité profonde avec signe de Romberg et perte de sensibilité
osseuse au diapason).
Examens biologiques
Anémie chronique à 4.9 g/dl macrocytaire arégénérative, taux de réticulocyte à
72700/mm³, plaquette à 39000/mm3. Leuco-neutropénie avec polynucléaires
à noyau hyper segmenté.
Augmentation de la sidérémie, de la bilirubine totale, libre et du LDH.
Myélogramme: aspect bleu, riche, érythroblastose à 24%, nombreuse mégaloblaste
avec corps de Jolly, parfois noyau irrégulier, polynucléaires de grande
taille, grosses plaquettes.
Présence d’AC anti facteur intrinsèque.
Achlorhydrie.
Examens histologiques
Biopsie gastrique : gastrite atrophique avec infiltration lympho plasmocytaire de la lamina propria.
Épreuve thérapeutique
Crise réticulocytaire au septième jour sous vitamine B12, taux de reticulocytes
à 1 M/mm³ et Hb: 7.6 g/dl
Avant traitement
L’évolution était marqué par :
Cliniquement :
Régression du syndrome anémique, régression plus au moins complète du syndrome neurologique.
Biologiquement:
en 72heures régression de la mégaloblastose, en 7 jours crise
réticulocytaire,
en 10 jours régression de la leucopénie, en 1 -2 mois disparition de l’anémie,
en dernier disparition de l’ hyper segmentation des PNN.
Après traitement
Cinq ans plus tard, elle est revenue pour une sécheresse buccale et une impression d’avoir du sable dans les yeux, très marquées ces derniers mois. Elle décrit une perte de poids de 10 Kg en deux mois avec perte d’appétit et une fatigue inhabituelle. L’examen clinique retrouve un syndrome sec oculaire et buccal, des poly arthralgies inflammatoires et un syndrome tumoral fait de multiples adénopathies superficielles cervicales, associé à une hépato-splénomégalie.
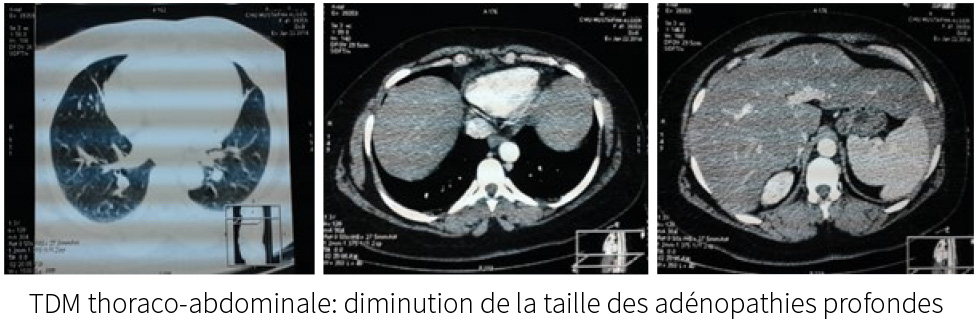
Le diagnostic d’un syndrome de Goujerot Sjögren est évoqué, sur:
La clinique :
Syndrome sec: xérostomie, xérophtalmie compliquée d’une kératite sèche
type
filamenteuse et ulcération de la cornée.
Parotidite bilatérale
Arthralgies inflammatoires (avec dérouillage matinal de 20 minutes) bilatérales,
symétriques, distales intéressant les métacarpo-phalangiennes, les inter-phalangiennes
proximales et sans signe d’érosion.
Syndrome tumoral.
La biologie :
Un syndrome inflammatoire avec hyper gammaglobulinémie polyclonale à
25g/l,
VS accélérée 135/145 mm.
Anticorps antinucléaires sont mouchetés à 1/1000 UI, AC anti SSA et anti SSB
positifs.
L’histologie :
Biopsie des glandes salivaires accessoires: infiltrat lympho plasmocytaire en amas significatif, stade 3 ou 4 de Chisholm.
Le syndrome tumoral fait :
Examen clinique Anorexie, amaigrissement de 10kg en deux mois.
De multiples adénopathies superficielles cervicales.
Hépatomégalie à bord inferieur ferme et sensible sans signes d’hypertension
portale.
Splénomégalie.
Examens biologiques
Syndrome inflammatoire: VS 135/145mm avec une hyper gamma globulinémie
à 25g/l, polyclonale, les IgG à 19 g/L (1,5xN), les IgM à 1.4 g/L (1,2xN) et les
IgA à 3.4 g/L (normale).
Pas de signe d’insuffisance hépatocellulaire.
Une cholestase anictérique: bilirubine totale à 08 μmol/L (N), PAL à 988 Ul/L
(8xN), ɣGT à 740 UI/L (20xN).
Une cytolyse légère: ASAT à 41 UI/L (1,3x N), ALAT à 43 UI/L, (1,3x N).
Examens morphologiques :
L’échographie cervicale : adénopathies sous maxillaires.
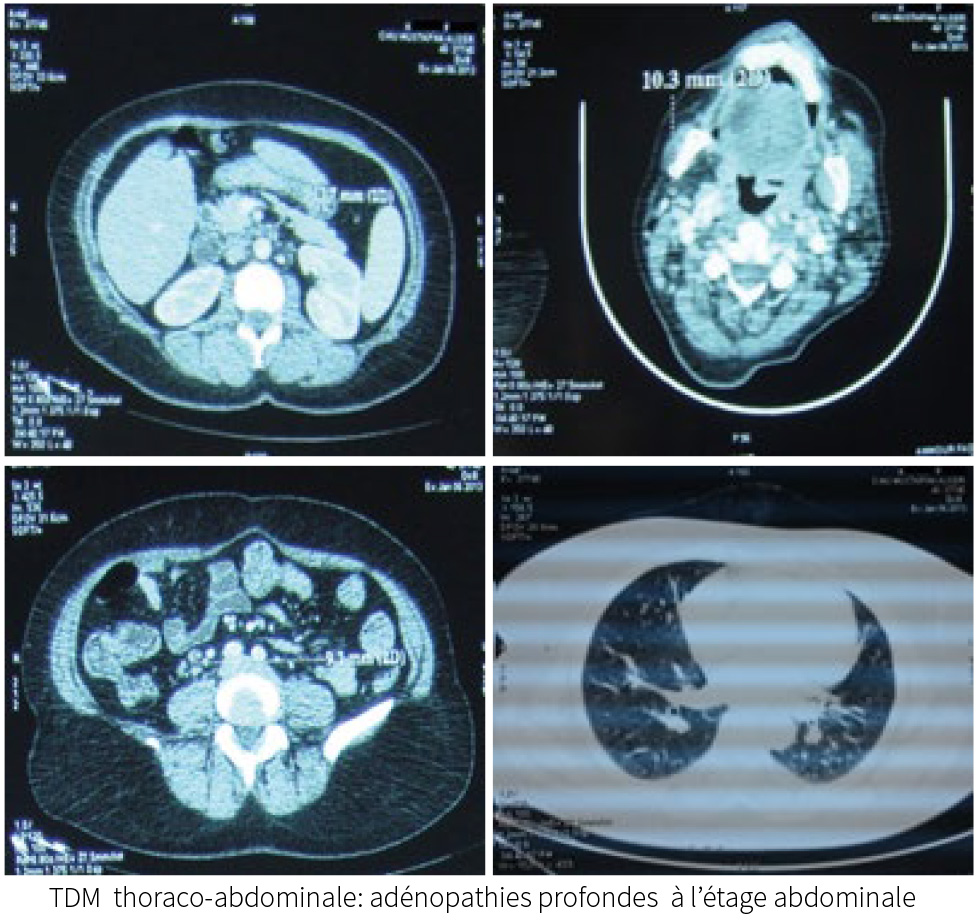
La TDM thoraco-abdominale:adénopathies coelio-mésentérique lombo-aortique
et inter aorto-cave avec hépato-splénomégalie.
Devant ce syndrome tumoral plusieurs diagnostiques ont été recherchés puis éliminer
Infections chroniques
Bactériennes:
Endocardite infectieuse : pas de souffle, échodoppler cardiaque normal
Sepsis : hémoculture négative
Abcès profond : absent
Tuberculose : pas de notion de contact ni signe d’imprégnation tuberculeuse,
recherche de BK aux crachats, tubage gastrique et urinaire négative ainsi que
l’IDR à la tuberculine à 7mm.
Syphilis : les sérologies sont négatives
Maladie de Whipple : pas de signes clinique ou infectieux
Virales (VIH, hépatites virales B, C, CMV, EBV) : sérologies négatives
Absence de syndrome mononucléosique.
Parasitose: toxoplasmose, paludisme, leishmaniose: sérologies négatives.
Hémopathies :
Syndrome lympho et myéloprolifératif :
Pas d’antécédent de thrombose veineuse ou artérielle, absence de syndrome
d’hyperviscosité, pas d’hyperglobulie, d’hyperleucocytose ou d’hyperplaquettose.
Leucémie lymphoïde chronique :
Terrain auto immun, syndrome tumoral
Frottis sanguin: globule rouge sans anomalie, globule blanc (60 02 00 98
02), richesse plaquettaire normal petite isolée
Syndrome hémophagocytaire :
Signes généraux : amaigrissement, altération de l’état général, adénomégalie
et hépato splénomégalie.
Syndrome inflammatoire et biologie hépatique perturbée.
Pas de signe hématologique de syndrome hémophagocytaire ni de troubles
d’hémostase.
Pas de dysfibrinogénémie ni d’hyper triglycéridémie ni d’ hyper férritinémie.
Absence d’hémophagocytose et d’éléments figurés par les macrophages
Transformation lympho proliférative d’un syndrome de Goujerot Sjögren :
Signes généraux: altération de l’état général, amaigrissement
Syndrome tumoral: adénopathies périphériques et hépato-splénomégalie
Examen ORL : bombement de la paroi postéro supérieur du cavum latéralisé à
droite à la naso fibroscopie.
La normalisation d’électrophorèse des protéines et d’immunofixation des Ig
Etude anatomopathologie: muqueuse naso-pharyngée d’aspect polypoïde,
épithélium métaplasique malpighien. Chorion lymphoïde avec persistance de
follicule à centre clair, sans évidence de cellules malignes.
Dégénérescence ou tumeur gastrique
Complication d’une anémie de Biermer évoluant depuis 05 ans : FOGD, ainsi que les biopsies étagées sont sans anomalies.
Le diagnostic d’hépatite auto-immune type 1, après avoir éliminé d’autres étiologies, est retenu devant :
Clinique
Amaigrissement
Biologique
Cholestase anictérique sans obstacle
Cytolyse hépatique (1,3xN)
Hyper IgG à 19 g/L (1,5xN),
AC antinucléaires élevé 1/1000 UI
AC anti muscle lisse positive à 1/ 80 UI (2xN).
Les AC anti mitochondrie, anti LKM1 sont négatifs.
Morphologique
L’échographie abdominale et l’examen par résonance magnétique nucléaire des voies biliaires (Bili-IRM) sont normaux, il n’existe pas de dilatation des voies biliaires intra ou extra-hépatiques.
Histologique
La biopsie hépatique a objectivé des lésions d’inflammation lympho plasmocytaire modérée péri portale. Il n’y a pas de granulome épithélioïde ou giganto- cellulaire.
Un traitement a été instauré selon les recommandations de l’AASLD: Prednisone (30 mg/j) avec dégression progressive pendant 05 semaines, combinées à l’Azathioprine (50 mg/j) et Plaquenil 2OO mg/j pour le syndrome de Gougerot Sjögren. L’évolution clinique était progressivement favorable (disparition de l’altération de l’état général, diminution du syndrome tumoral).
DISCUSSION
Les syndromes auto-immuns multiples constituent un cadre nosologique à ne pas méconnaitre (2, 4,6). La survenue simultanée d’une hépatopathie auto immune, d’un syndrome de Gougerot Sjörgen et d’une thyroïdite auto immune n’est certainement pas fortuite (1, 5,7). II existe en effet de nombreuses similitudes en ce qui concerne les facteurs épidémiologiques, génétiques, histologiques et immunologiques(4,6). Leur classification est marquée par l’association chez un même patient d’au moins trois maladies auto-immunes spécifiques et non spécifiques d’organes (2, 6, 7). Ces SAIM sont de fréquence rare comme en témoignent les publications de cas isolés et traduisent un trouble de régulation de la réponse immunitaire sur un terrain génétique particulier (1, 7). Les points essentiels à retenir sont:
- la recherche systématique d’une atteinte thyroïdienne avec dosage des anticorps antithyroïdiens. En cas de positivité, les autres maladies auto-immunes devront être recherchées tous les deux ans dans un intérêt diagnostic et thérapeutique (2, 3, 6).
- Le SAIM traduit une sensibilité individuelle majeure à la perte de la tolérance immunologique des antigènes du soi. La fréquence des maladies auto- immunes est plus importante chez les apparentés au 1er degré d’un patient atteint de SAIM en comparaison des groupes contrôle (2, 4).
- En cas d’hépatopathie auto-immune, l’association à d’autres maladies dysimmunitaires extra hépatiques est rapportée dans 20% des cas (7), incite à leur recherche systématique dans un but de diagnostic et d’un traitement précoce.
- Le syndrome de Gougerot Sjören qui apparait au deuxième rang est étroitement corrélé à la thyroïdite auto-immune (3, 5), ce qui suggère I’ existence d’un ou plusieurs facteurs étiologiques communs.
- Enfin, le syndrome des anticorps anti-phospholipides vient compléter le tableau (chez les deux cas présentés on ne note pas de signes obstétricaux, d’antécédents thrombophlébitiques et le bilan de SAPL est revenu négatif).
CONCLUSION:
L’identification des nouvelles formes d’association des maladies auto immunes soulève de nombreuses questions physiopathologiques, qui devraient conduire à une meilleure connaissance des facteurs génétiques de susceptibilité à l’ origine de ces maladies. Les anticorps font partie des critères diagnostiques, leur interprétation doit être en fonction du contexte clinique, des données biologiques, morphologiques et histologiques.
BIBLIOGRAPHIE
- A. Zulfiqar, J.-L. Pennaforte, M. Drame, E. Andres. La maladie de Biermer au cours des syndromes auto-immuns multiples: étude rétrospective de 74 observations et revue de littérature; Revue de Médecine Interne décembre 2012 ; Vol 33 - N° S2P. A104-A105.
- M. Michaud, C. Bonis, C. Conor, A. Inchauspe, S. Broussaud, C. Couteau, M.J. Ferro, S. Fontaine, D. Garipuy, F. Gaches. Syndrome auto-immun multiple: à propos d’une série de huit cas; Revue de Médecine Interne décembre 2012 ; Vol 33 - N° S2P. P. A108-A109.
- W. Chebbi, O. Berriche, B. Zantour , W. Alaya, M.H. Sfar. Pathologie thyroïdienne et syndromes auto-immuns multiples: à propos de 10 observations; Revue de Médecine Interne décembre 2012; Vol 73 - N° 4 P. 328.
- B.Bonnotte, JL Dupond, K Boucho, H Roussef. Physiopathologie des maladies auto immunes; Revue de Médecine Interne 25 (2004) ; 648- 658.
- S. Humbert, A. Le Quellec, H. Gil, N. Méaux- Ruault, N. Magy-Bertrand. Différences clinico-biologiques entre les syndromes de Gougerot–Sjögren isolés et ceux associés à d’autres maladies auto-immunes : étude bicentrique de 206 patients ; Revue de Médecine Interne juin 2013 ; Vol 34 - N° S1 P. A61-A62.
- A. Zulfiqar, M. Drame, J.L. Pennaforte, E. Andres. Fréquence des maladies auto-immunes chez 188 patients atteints de la maladie de Biermer ; Revue de Médecine Interne décembre 2012 ; Vol 33 - N° S2 P. A105-A106.
- N. Maamouri , H. Kchir , A. Mebazza , I. Ben Nacef , N. Belkahla , H. Ouerghi , F. Ben Hariz , S. Chouaib , H. Chaabouni , A. Ben Osman , N. Ben Mami . L’atteinte hépatique au cours du syndrome auto-immun multiple : à propos de 15 cas ; Revue de Médecine Interne juin 2010 ; Vol 31 - N° S1. P. S167.